Perinatal Software:
An Updated Primer on the Physiology of Fetal Heart Rate Control
Understanding how the heart reacts to labor is essential knowledge for clinicians who are responsible for the interpretation of fetal heart rate tracings
by Emily Hamilton, MDCM and Philip Warrick, Ph.D.
Animal labs provide an opportunity to measure exactly what is happening internally as external conditions are manipulated. Better instrumentation has improved the measurement of physiological responses in animal experiments and electronic databases have increased our capacity to test hypothesis on tracings collected from human births.
Early physiological experimentation concluded that hypertension was a key component initiating decelerations by stimulating the baroreceptors with consequent FHR slowing via efferents in the vagus nerve. More recent work provides a simpler proposal namely that the chemoreflex with both parasympathetic and sympathetic components is the major initiator of fetal heart rate decelerations. In this proposal the chemoreflex efferent in the vagus nerve causes FHR slowing and the sympathetic component causes hypertension to maintain perfusion to vital organs.
The following review is by no mean an exhaustive discussion of the thousands of published articles on fetal heart rate monitoring. We have attempted to produce a succinct review the physiology of fetal heart rate control that is relevant to the clinician caring for women in labor. In conclusion, apart from unrelenting bradycardia, there is no signal FHR marker that consistently indicates serious fetal compromise. Likewise, there is no single marker that reliably excludes metabolic acidemia. The frequency and size of decelerations are important indicators as well as the nature of the FHR between contractions and trends over time.
The clinical goal of electronic fetal monitoring is to identify fetuses with increased risk of hypoxic injury so that intervention can avoid adverse outcome without also causing excessive numbers of unnecessary interventions. Understanding the mechanisms of fetal heart rate control are important because they help us to infer the physiological state of the baby and gauge whether intervention is really necessary. While this summary provides the basics for understanding heart rate regulation, it is important to remember that our understanding of this physiology continues to evolve.
The fetal heart rate results from the combined effect of several fluctuating influences mediated through extrinsic and intrinsic cardiac control mechanisms.
In adult cardiology, ECG changes are very helpful to diagnose myocardial infarction because there is a direct relationship, precisely defined by anatomy and electromagnetic physics, linking the presence of Q waves in the 12-lead ECG to the location of injured myocardium. In fetal surveillance, the situation is very different. Standard monitoring provides only the heart rate, not the ECG. Clinicians depend upon the heart rate to infer the condition of another organ, namely the fetal brain. Although the fetal heart rate is related to fetal brain state, it is also affected by a number of other factors. The fetal heart rate results from the combined effect of several fluctuating influences mediated through extrinsic cardiac control mechanisms (sympathetic and parasympathetic nerves and circulating catecholamines), as well as intrinsic cardiac factors (myocardium and its conducting system). The following text describes the principal physiological pathways that control heart rate and influence heart rate variability.
The heart is a muscle with its own pacemaker, conducting system, numerous types of receptors (alpha and beta adrenergic) and direct neuronal connections to both the sympathetic and parasympathetic systems. Ultimately, any influence on heart rate is mediated by one or more of these structures. The basic anatomy and physiology of heart rate control are described in textbooks on physiology and fetal monitoring.1,2 The schematic diagram shown in Figure 1 summarizes the heart rate control pathways.
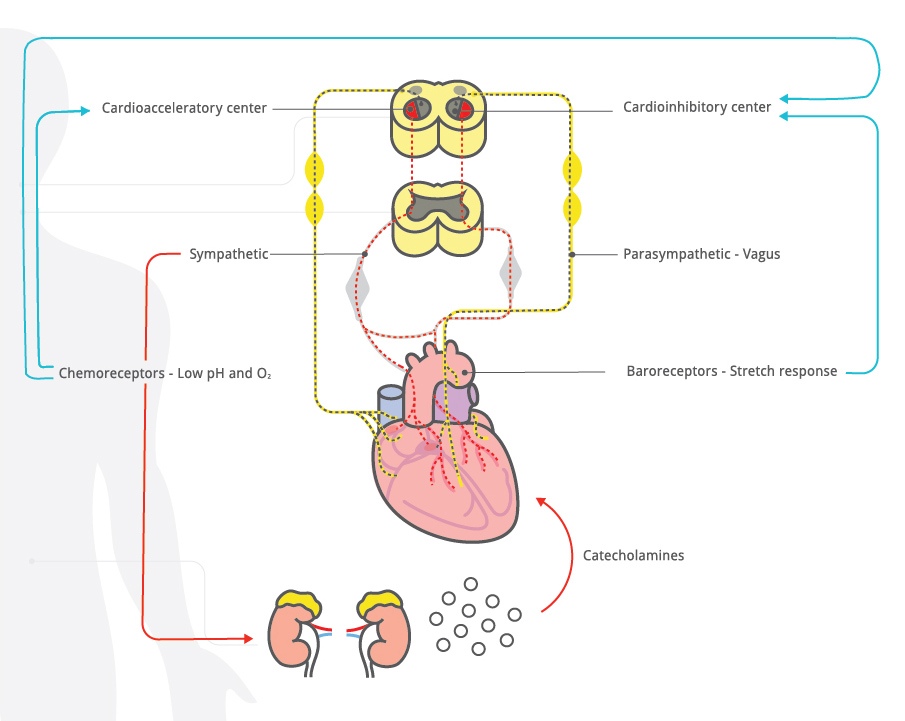
Figure 1: Schematic outline of heart rate control mechanisms.
The cardioregulatory center in the medulla oblongata contains an acceleratory center and an inhibitory center. The cardioregulatory center receives input from the central nervous system, reflex pathways and circulating catecholamines. An example of central nervous system influence on the acceleratory response is seen with vibroacoustic stimulation. In response to sudden auditory stimulation, the central nervous system activates the cardioacceleratory center. The cardioacceleratory center increases heart rate directly via sympathetic cardiac nerves which interact with the sinoatrial node to increase the heart rate. The cardioinhibitory center slows the heart rate via the parasympathetic vagus nerve which can slow heart rate by modulation at various levels, including the sino-atrial node. Reducing cardioinhibitory activity increases heart rate.
The rapidity of heart rate change is determined by the conditions that trigger the change.
Arterial baroreceptors are sensitive to stretch or distension of a vessel caused by blood pressure changes. An increase in arterial blood pressure produces vessel distension and causes arterial baroreceptors to send neuronal messages to the cardioinhibitory center, which in turn causes rapid slowing of the fetal heart rate via the parasympathetic vagus nerve.
Physiologists continue to elucidate the important role of fetal chemoreceptors and their effect on fetal heart rate. 3-15 Chemoreceptors are found in peripheral locations such as in the aortic arch and carotid arteries where they are sensitive to low oxygen levels. 7 Chemoreceptors located in the brainstem are sensitive to low pH and elevated pCO2. In neonatal life activation of these centers induces strong stimulation for breathing. In fetal life, pulmonary ventilation does not occur and is not a source of fetal oxygenation. The chemoreceptor mediated ventillatory response is inhibited in fetal life and other components of the chemoreflex responses are more apparent.8
A prominent CNS response to fetal chemoreceptor stimulation is increased sympathetic activity, inducing peripheral vasoconstriction and favoring preferential blood flow to vital organs. A second component of the chemoreflex response to hypoxemia is parasympathetic, mediated via the vagus nerve, resulting in a fall in heart rate. This effect is believed to benefit the fetus by temporarily reducing myocardial work to compensate for diminished oxygen availability.3-6 A third consequence of chemoreceptor activation of the sympathetic system causes release of other substances such as adrenal catecholamines and vasopressin.
The catecholamines, epinephrine and norepinephrine, secreted from the adrenal, are both hormones and neurotransmitters. Norepinephrine binds to beta receptors in the heart causing an increase in heart rate, contractility and stroke volume. Catecholamines can also cause redistribution of blood flow by inducing vasoconstriction and vasodilation in different regions. Vasoconstriction is mediated through the α-adrenergic receptors in liver, kidney, skin and gut, and vasodilation is mediated through β adrenergic receptors in skeletal muscle. Catecholamine release, stimulated by the sympathetic nervous system may be precipitated by stress conditions, such as loud sounds, fear or low blood sugar as well as low oxygen and pH levels or elevated pCO2 levels.
The overarching mission of the cardiovascular system is to deliver sufficient oxygen to key organs… Any event that perturbs the fetal cardiovascular system will initiate a cascade of compensatory actions to optimize oxygen delivery and restore fetal hemostasis.
The medulla oblongata also contains the vasomotor center that regulates peripheral blood vessel dilation and constriction to help maintain normal blood pressure and distribute blood to vital organs. It also responds to baroreceptors, chemoreceptors and catecholamines.
The rapidity of heart rate change is related to the rapidity of the conditions that trigger the change. Central stimuli like a sudden loud sound or a quick increase in blood pressure cause rapid heart rate changes mediated by direct neuronal pathways to the sinoatrial node. Catecholamine mediated effects are relatively slow reflecting their half-life of 2 to 3 minutes.
While all of the mechanisms described above modulate heart rate, it is important to underline that the overarching mission of the cardiovascular system is to deliver sufficient oxygen to key organs. Changing heart rate is an important component of this mission but not the only one. Other cardiovascular compensatory mechanisms include changes in blood pressure, cardiac stroke volume, local vasoconstriction or vasodilation to promote redistribution of blood flow and alterations in hemoglobin-oxygen dissociation to increase oxygen extraction. Any event that perturbs the fetal cardiovascular system will initiate a cascade of compensatory actions to optimize oxygen delivery and restore fetal hemostasis. The fetal heart rate pattern we observe is the combined results of many physiological actions.
Fetal Heart Rate Decelerations
For a concise review of the historical experimental work elucidating the mechanisms of fetal heart rate decelerations and response to hypoxemia, the reader is referred to excellent descriptions by Freeman et al and 2012 and Martin et al 2008 2,9
Cord compression was simulated by constricting devices placed on the cord.
Umbilical cord compression has been created in research environments by actually compressing the fetal cord with implanted mechanical devices that could be manipulated to compress the cord to different degrees and durations. Decreased uteroplacental oxygen delivery was generally simulated by compressing the arteries supplying blood to the uterus or controlling the levels inspired of oxygen. Pharmacological blocking agents were added to inhibit the effects of the sympathetic and/or parasympathetic systems. Some experiments even used vagotomy to eliminate the effects of the vagal system. Inserted sensors measured blood pressure, heart rate and acid base status during the experiments.
Uteroplacental dysfunction was simulated by compressing blood flood to the uterus or decreasing inspired 02 levels.
Variable Fetal Heart Rate Decelerations
Classic teaching on the mechanisms of variable decelerations is outlined in Figure 2. 2 In this physiological model, the final steps in two pathways involve hypertension produced directly by umbilical artery obstruction or indirectly by peripheral vasoconstriction. The pathway on the left was thought to be initiated during umbilical cord compression that was not accompanied by hypoxemia or pH changes. Hypertension developed in response to compression of the umbilical arteries. In turn, the stretch-sensitive baroreceptors were activated, stimulating the cardioinhibitory center and causing rapid slowing of the heart rate mediated via the vagus nerve. Release of the cord compression relieved the hypertension, reduced cardioinhibitory center activity which reduced vagal stimulation and the fetal heart rate rose rapidly. 10
The second column outlines pathways thought to be present when cord compression is accompanied by hypoxemia and acidemia. Here low pH and hypoxemia trigger the chemoreceptor response causing peripheral vasoconstriction. The resulting hypertension induces the barorflex, stimulating the cardioinhibitory center and its vagal efferent to slow the heart rate.
The last column shows an additional mechanism thought to be present with marked fetal acidemia where the heart rate slows due to direct myocardial effects. Evidence of a non-vagal (intrinsic myocardial) source of deceleration was demonstrated in experiments with complete vagotomy. Fetal heart rate deceleration, albeit somewhat delayed, still occurred in response to cord compression. 6, 11

Figure 2: Three proposed pathways with variable decelerations, adapted from Freeman et al(2)
Classic teaching on the mechanisms of Late decelerations is outlined in Figure 3.
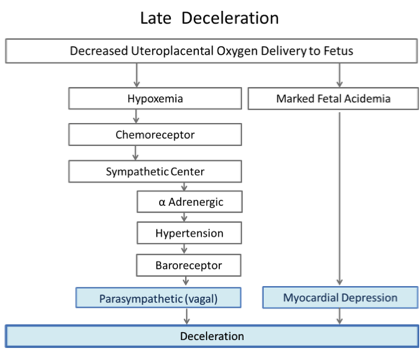
Figure 3: Two proposed pathways with late decelerations, adapted from Freeman et al. (2)
In this physiological model, the first pathway also involves hypertension. To simulate decreased uteroplacental oxygen delivery, they applied repeated occlusions to the maternal hypogastric artery12. The occlusions resulted in fetal hypertension and vagally mediated decelerations. The degree of hypertension and the amount of deceleration were closely related, although some deceleration remained when the transient hypertension was prevented by α adrenergic blockade. The timing of the onset, nadir and end of the deceleration was delayed with respect to the occlusion and mirrored the timeline of the hypertensive response. Vagal blockade eliminated these decelerations in the non-acidemic sheep. Thus, “intermittent placental insufficiency” can cause decelerations. In this model hypertension followed by baroreceptor response were proposed as the key steps stimulating the cardioinhibitory center and causing slowing of the heart rate mediated via the vagus. These “late” decelerations were not associated with fetal acidosis.
With progressive acidemia the decelerations became deeper and longer. In the presence of very severe acidemia (pH=6.96) they could not be eliminated by vagal blockade. With complete vagal and α and β adrenergic blockade, the decelerations persisted. The fetal heart, devoid of any sympathetic and parasympathetic influences, showed decelerations suggesting that intrinsic myocardial depression was the deceleration mechanism in the presence of severe acidosis and hypoxia.12
Even in the sheep experiments using precisely controlled conditions, consistent fetal heart rate decelerations could not be produced equally in all animals despite 2 hours of repetitive maternal vascular occlusions.12 The “late” decelerations induced by constricting the sheep uterine arteries with and without acidemia are redrawn to scale on the grid used by fetal monitoring paper in Figure 4.
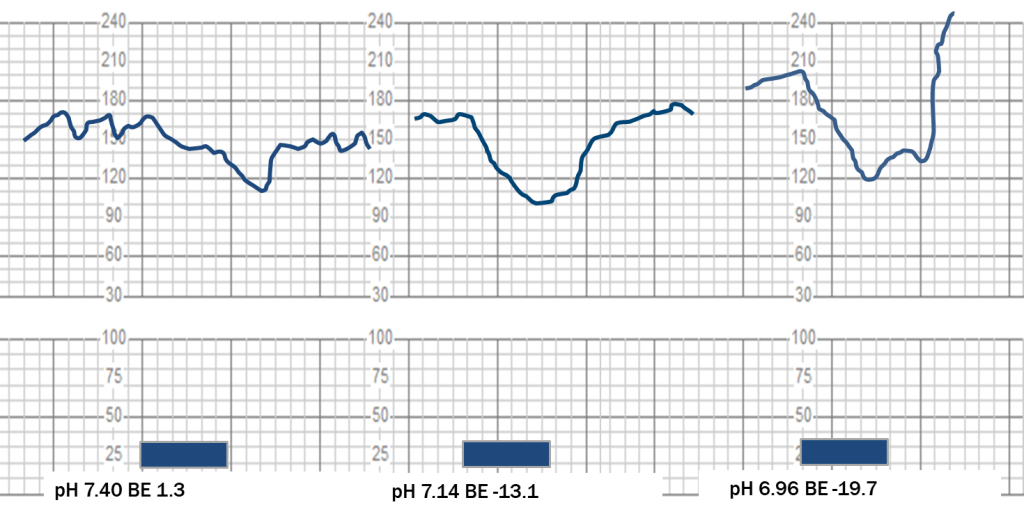
Figure 4: Decelerations induced by 1 minute hypogastric artery compression with and without acidemia redrawn to scale on fetal monitoring paper grids, adapted from Martin et al. (12)
Over that last decade mounting evidence based on recent animal experimentation with very sensitive sensors and finer time resolution, has produced more information about hand chemoreceptors and their role in the production of decelerations. Some authors have challenged the role of the baroreceptor response as the sole or major cause of fetal heart rate decelerations.6, 13-15 Clinicians who use fetal heart rate recording are strongly encouraged to read in detail excellent summaries of their studies on the mechanisms of fetal heart rate decelerations.6, 13-15
In one set of sheep experiments, blood pressure and fetal cerebral oxygenated hemoglobin were measured during complete cord compression. Fetal heart rates decelerated rapidly and well before sustained hypertension developed. Decelerations more closely followed cerebral oxygenation. After 2 minutes of complete cord compression, the FHR in the deceleration began to recover despite continuing fall in cerebral oxygenation and rising hypertension. Clearly other compensatory mechanisms intervened.14
Repetitive complete cord compressions are not a natural physiological condition. Experiments were designed with less drastic interventions to simulate more realistic clinical conditions. In another, set of experiments with sheep in labor, uterine contractions were associated with decelerations. Internal sensors showed that the contractions were associated with an initial fall in mean arterial pressure and an abrupt fall in the FHR. Recovery of the FHR was seen as mean arterial pressure rose. Thus the opinion that most decelerations are purely baroreceptor mediated was not supported by these recent studies.14, 15
Other work by Bennet et al has also challenged common teaching about the relationship between hypertension and decelerations. In a series of sheep experiments they applied 60 second complete cord compressions every 5 minutes in one group and an identical cord compression every 2.5 minutes in a second group. The sheep in the first group maintained normal gases and all sheep in the second group developed severe acidemia.13 Deep and wide decelerations with an abrupt onset were present with all cord compressions. Decelerations were present when there was mild hypertension during the compression but they were also present when marked hypotension accompanied the compression. With advanced acidemia and hypotension, decelerations were even deeper and steeper. The nadir of the deceleration was very close to the end of the compression period in both groups. The FHR returned to predeceleration levels after the compressions ended in both groups. Recovery time took about 30 seconds longer in the most acidemic set. This experiment also demonstrated that adequate time between cord compressions “reperfusion time” was essential to maintain normal acid base status.
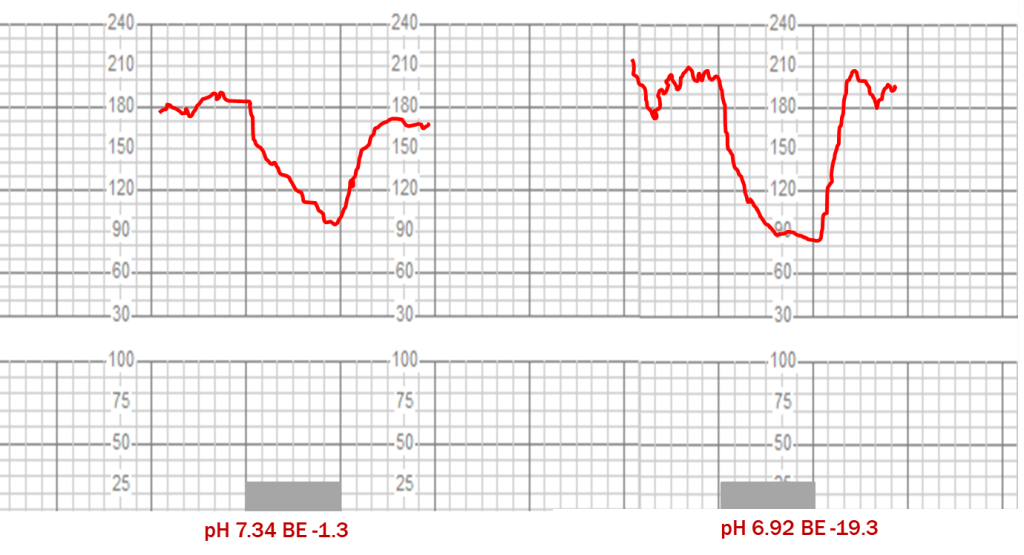
Figure 5: Decelerations induced by complete one minute cord compressions with and without acidemia redrawn to scale on fetal monitoring paper grids, adapted from Bennet et al (23)
In Figure 6, the decelerations from these two different mechanisms are superimposed demonstrating how size, shape and timing tend to converge in the presence of marked acidemia irrespective of the initial event precipitating the deceleration.

Figure 6: Superimposed decelerations from uterine artery compression (in blue) and cord compression (in red).
Based on their experiments in sheep, this group of fetal physiologists has proposed a different sequence of events to explain decelerations: namely contractions induce a reduction in uteroplacental blood flow and/or cord compression causing a reduction in fetal pO2 that triggers the chemoreflex whose magnitude is related to the degree of hypoxia. 6, 13-15 The parasympathetic component of the chemoreflex causes a rapid decrease in heart rate to lessen cardiac work and its oxygen need. The sympathetic component induces peripheral vasoconstriction and hypertension to maintain perfusion and adequate oxygen delivery to vital organs. Provided the repetition of these episodes was relatively infrequent, the healthy fetus could adapt for many hours. Factors that modulate fetal anaerobic reserve are myocardial glycogen stores, placental exchange capacity and chronic hypoxia. Factors that contribute to progressive oxygen debt are strength, duration and frequency of decelerations. Fetal compensation or decompensation is a function of all of these factors. Their proposed pathways with contraction related decelerations are illustrated in Figure 7.
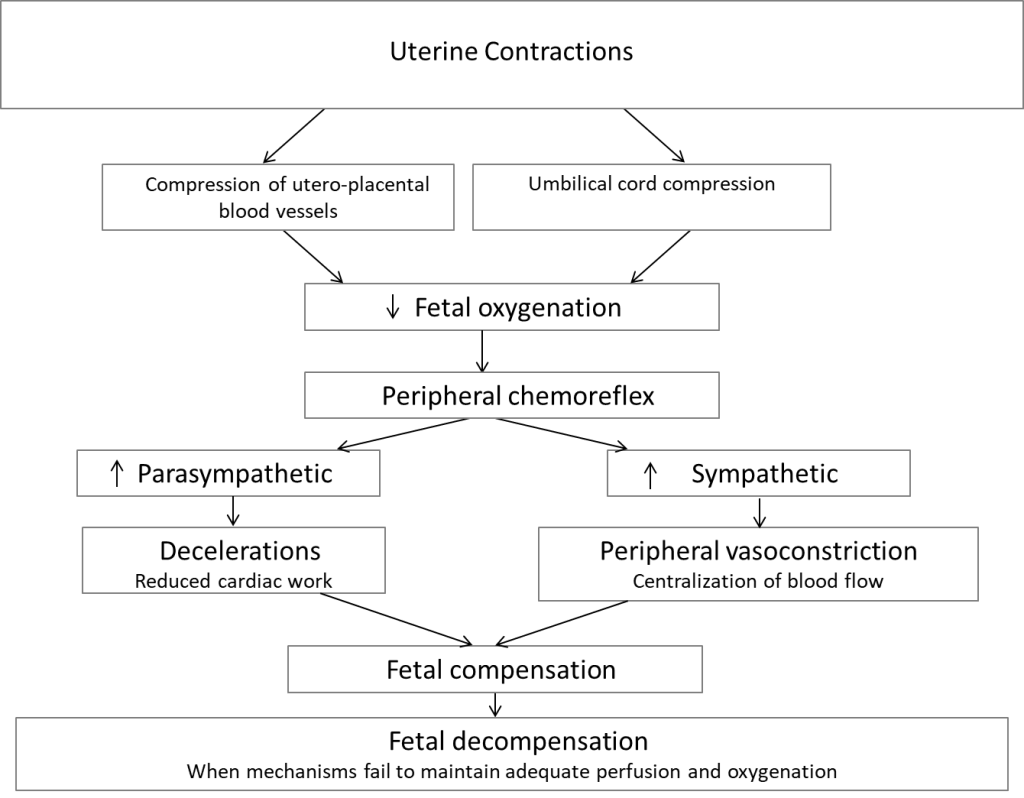
Figure 7: Proposed mechanisms of intrapartum deceleration, adapted from Lear et al. (13) and Bennet et al. (14)
Shallow gradual deceleration that are late in timing and accompanied by a normal baseline and moderate baseline variability are inconsistent markers of low Apgar scores or metabolic acidosis. The late timing reflects the lag between insult onset (the contraction) and actual reduction in fetal oxygenation. The fetus is exposed to the lower intervillous oxygen levels only when the uterine contraction forces are sufficient to impede intramyometrial perfusion. Maternal perfusion in the intervillous space is generally maintained during the early part of a uterine contraction. They suggested that late decelerations tend to occur in a fetus with limited reserves that is exposed to modest reductions in uterine blood flow that would not cause bradycardia in a healthy fetus. These opinions on late decelerations are consistent with human observations by Sameshima et al who reported that that acidemia was present at birth in only 20% of babies with recurrent late decelerations and loss of variability. Acidemia was most frequent, (42%) but still not inevitable when recurrent late decelerations and minimal/absent variability were present on admission.16
Fetal Oximetry and Fetal Heart Rate Decelerations
Fetal oximetry experience preceding and during the FOX trial has also provided some useful data on conditions associated with fetal heart rate decelerations. The degree of oxygen saturation of fetal hemoglobin can be estimated based on the different absorption of red and near infrared light by oxygenated and deoxygenated fetal hemoglobin. Sensors placed against the fetal cheek measure the reflected light in two specific wavelengths and from this the percentage of fetal oxygen saturation can be estimated. Using fetal oximetry probes many centers reported images of deep decelerations with little or no change in fetal oxygen saturation. Assuming that the oximetry sensors are accurate, these findings demonstrate that deep decelerations can occur in the absence of significant peripheral hypoxemia.17
Heart Rate Variability
All of the mechanisms controlling fetal heart rate depicted in Figure 1 influence heart rate variability.17-22 Fetal behavioral states, breathing and movements affect heart rate variability acting though the central pathways to the medulla, and then to the heart via the sympathetic and parasympathetic systems. Fetal heart rate variability is suppressed by factors that depress fetal brain function. Circulating catecholamines increase heart rate variability.18
Animal experiments have shown that blockage of the parasympathetic system with atropine results in a reduction in short-term variability. Some authors but not all have shown that a reduction in long-term variability occurs after sympathetic blockade.19-22 Fetal heart rate variability is more than the simple “push-pull” interactions between the inhibitory and acceleratory limbs of the autonomic nervous system. The heart itself contributes to variability. Even with complete pharmacologic blockage of the sympathetic and parasympathetic systems, around 35-40% of fetal lamb heart rate variability persists.19
The association between variability and metabolic acidosis is less clear. This association is important because all contemporary EFM classification methods place high reliance upon baseline fetal heart rate variability to exclude the presence of metabolic acidosis.23-30 The 2008 NICHD Update publication in which the Category I, II, III classification method was first described includes a statement that “moderate variability reliably predicts the absence of metabolic acidemia at the time that it is observed”.24 This concept was softened in the 2009 ACOG Practice Bulletin 106 with the statement “The data relating FHR variability to clinical outcomes, however, are sparse.”25 This practice bulletin endorsed the 3-level categorization of tracings where the third level required absent baseline variability. None of these guidelines specify what level of metabolic acidemia or degree of reliability they mean.
The body of literature, that disputes the statement that moderate variability reliably excludes the presence of metabolic acidemia, continues to grow. In animal studies, Martin demonstrated that in sheep the initial fetal heart rate response to sudden hypoxemia was a slowing of the heart rate with increased variability.12 Others observed similar changes in sheep and in monkeys.31-33 Field et al found initial decreases in heart rate variability with iliac occlusion in sheep, but variability returned to normal by 36 minutes despite worsening metabolic acidosis.34 These observations of normal variability in the face of acidemia led researchers to postulate that some aspect of variability control could be different in animals compared to humans.
Several reports based on observations in human births do not support the reliability of moderate baseline variability to excluded acidemia. In the human literature, four recent and independent studies using various definitions of acidosis and examining the last 30-60 minutes of the tracing reported that the percentage of babies with acidosis who had moderate variability ranged from 15% to 91%.35-40 Even with near lethal levels of uterine artery base deficit (>=16 mmol/L), a full 15 to 32% of fetal tracings had moderate baseline variability in the portion recorded just before birth.35 In a recent expert review of 120 cases of term babies born with base deficit >12mmol/L levels only 13 % showed minimal or absent variability.39 Most recently a review of fetal heart rate patterns in term babies with a uterine artery pH >7.1 found the most predictive FHR parameters were cumulative deceleration area with any amount of tachycardia. The presence or absence of moderate variability did not modify the relationship even when moderate variability was always present.40
Transient loss of variability usually is associated with benign conditions like fetal sleep or maternal medications. Persistent low variability is concerning, especially when present on admission or associated with decelerations and tachycardia.16 Low variability must not be ignored, but it appears that moderate variability does not guarantee the absence of acidemia.
How Do These Discoveries Affect the Clinical Interpretation of Tracings?
Collectively animal experiments have demonstrated the following facts about decelerations:
- Cord compression and uterine contractions can induce decelerations.
- Both the baroreflex and the chemoreflex have parasympathetic efferents in the vagus nerve.
- FHR decelerations can be associated with fetal hypertension or fetal hypotension. Hypotension indicates failing fetal compensation.
- FHR decelerations can be associated with little or no change in peripheral hemoglobin oxygen saturation.
- Fetal hypoxia can induce decelerations
- The size, shape and timing of decelerations induced by cord compression or uterine artery compression tend to converge in the presence of marked fetal acidemia.
In sum, these facts mean that it will always be difficult to draw conclusions about the cause of a deep and wide deceleration by inspecting only its size, shape and timing especially in the presence of acidemia. Cord compression does cause rapid fetal heart rate changes but so does a rapid change in pO2 and both of these mechanisms can produce their effect on the heart via the vagus nerve. Decelerations from cord compression can lead to severe hypoxemia and decelerations from uteroplacental insufficiency can be present without acidemia. In short, there is no single fetal heart rate parameter that consistently identifies fetal compromise. The frequency and size of deceleration are important indicators as well as the nature of the FHR between contractions and trends over time.
Apart from unrelenting bradycardia, there is no signal FHR marker that consistently indicates serious fetal compromise.
In practice, clinicians must integrate clinical data with electronic fetal monitoring patterns in order to project what is likely to happen and how quickly it might occur. As discussed throughout this article the relationship between heart rate and the underlying fetal condition is complex. The clinician’s task is not simple. In fact, one could consider that the substantial reduction the rates of intrapartum-related neonatal encephalopathy seen in numerous regions around the world demonstrate a remarkable achievement. Several large jurisdictions with population-wide statistics have shown steady declines, often reaching reductions in the range of 40% per decade. 41-49 One can speculate on some reasons. Little has changed in the physical devices that record fetal heart rate and contractions. On the other hand, clinical behavior has changed considerably. We have an increased understanding of fetal physiology, the natural evolution of fetal acidosis and the consequences of delayed intervention which in turn have led to better clinical practices. In-house obstetrical coverage, drills to increase the rapidity of emergency cesarean, safer oxytocin management protocols also contribute to better outcomes.
We are also entering a new chapter in the story of fetal surveillance made possible by the coexistence of 4 factors:
- Electronic storage of vast amounts of clinical data and tracings makes it feasible to study rare outcomes like severe metabolic acidemia or hypoxic ischemic encephalopathy.
- Automated analysis means that these digital tracing can be analyzed in a consistent quantitative fashion, not only for the classic patterns defined by the NICHD but for patterns and relationships that are not readily visible to the human eye.
- Fetal monitors are better at separating maternal and fetal heart rate.
- The final exciting element in the mix is the exceptional advance in Machine Learning. Machine Learning refers to a family of software algorithms designed to find the most predictive constellations of factors for a specific outcome by examining actual data. That is, the machine (computer) learns from the actual data. These methods are not held to a specific clinical hypothesis. They do not make assumptions about linear relationships or require independent effects and they can consider trends over time. They may detect several clusters or subgroups that are associated with the adverse outcome and provide an estimated probability of that occurrence.
Given the complexity of the underlying physiology controlling heart rate, the many clinical conditions that can wax and wane to affect oxygen delivery to the fetus and natural variation in fetal reserves, a mathematical method capable of determining the most predictive fetal heart rate patterns in specific clinical circumstances is welcome.
We have become accustomed the term a “Perfect Storm” when a collection of adverse conditions converge to produce calamity. In contrast, we are entering a stretch of “Perfect Weather” with the convergence of conditions collectively conducive to advancing the science of electronic fetal heart rate interpretation in actual clinical practice.
- Martini FH, Nath JL, Batholomew EF. Anatomy and Physiology. 9th ed. Boston, Benjamin Cummings;2012
- Freeman RK, Garite TJ, Nageotte MP, Miller LA. Fetal Heart Rate Monitoring. 4th ed. Philadelphia, PA: Lippincott, Williams and Wilkins; 2012
- Giussani DA, Spencer JA, Moore PJ, Bennet L, Hanson MA. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol. 1993 Feb;461:431-49.
- Fletcher AJ, Gardner DS, Edwards CM, Fowden AL, Giussani DA. Development of the ovine fetal cardiovascular defense to hypoxemia towards full term. Am J Physiol Heart Circ Physiol. 2006 Dec;291(6):H3023-34.
- Baan J Jr, Boekkooi PF, Teitel DF, Rudolph AM. Heart rate fall during acute hypoxemia: a measure of chemoreceptor response in fetal sheep. J Dev Physiol. 1993;19:105-11.
- Westgate JA, Wibbens B, Bennet L, Wassink G, Parer JT, Gunn AJ. The intrapartum deceleration in center stage: a physiologic approach to the interpretation of fetal heart rate changes in labor. Am J Obstet Gynecol. 2007 Sep;197(3):236.e1-11.
- Garabedian C, De Jonckheere J, Butruille L, Deruelle P, Storme L, Houfflin-Debarge V. Understanding fetal physiology and second line monitoring during labor. J Gynecol Obstet Hum Reprod. 2017 Feb;46(2):113-117
- Teitel DF. Fetal chemoreception: a developing story. Reprod Fertil Dev. 1996;8(3):471-82
- Martin CB Jr. Normal fetal physiology and behavior, and adaptive responses with hypoxemia. Semin Perinatol. 2008 Aug;32(4):239-42.
- Lee CY, Di Loreto PC, O’Lane JM. A study of fetal heart rate acceleration patterns. Obstet Gynecol. 1975 Feb;45(2):142-6
- Barcroft J. Researches on prenatal life. Blackwell Scientific Publications, Oxford, 1946
- Martin CB Jr, de Haan J, van der Wildt B, Jongsma HW, Dieleman A, Arts TH. Mechanisms of late decelerations in the fetal heart rate. A study with autonomic blocking agents in fetal lambs. Eur J Obstet Gynecol Reprod Biol. 1979 Dec;9(6):361-73.
- Bennet L, Westgate JA, Liu YC, Wassink G, Gunn AJ. Fetal acidosis and hypotension during repeated umbilical cord occlusions are associated with enhanced chemoreflex responses in near-term fetal sheep. J Appl Physiol (1985). 2005 Oct;99(4):1477-82
- Lear CA, Galinsky R, Wassink G, Yamaguchi K, Davidson JO, Westgate JA, Bennet L, Gunn AJ. The myths and physiology surrounding intrapartum decelerations: the critical role of the peripheral chemoreflex. J Physiol. 2016 Sep 1;594(17):4711-25
- Bennet L, Gunn AJ. The fetal heart rate response to hypoxia: insights from animal models. Clin Perinatol. 2009 Sep;36(3):655-72. doi: 10.1016/j.clp.2009.06.009.
- Sameshima H, Ikenoue T, Ikeda T, Kamitomo M, Ibara S. Unselected low-risk pregnancies and the effect of continuous intrapartum fetal heart rate monitoring on umbilical blood gases and cerebral palsy. Am J Obstet Gynecol. 2004 Jan;190(1):118-23.
- Freeman RK, Garite TJ, Nageotte MP. Fetal Pulse Oximetry in Fetal Heart Rate Monitoring. 3rd ed. Philadelphia, PA: Lippincott, Williams and Wilkins; 2013
- Bistoletti P, Lagercrantz H, Lunell NO. Fetal plasma catecholamine concentrations and fetal heart-rate variability during first stage of labour. Br J Obstet Gynaecol. 1983 Jan; 90(1):11-5.
- Dalton KJ, Dawes GS, Patrick JE. The autonomic nervous system and fetal heart rate variability. Am J Obstet Gynecol. 1983 Jun 15;146(4):456-62
- Wakatsuki A, Murata Y, Ninomiya Y, Masaoka N, Tyner JG, Kutty KK. Autonomic nervous system regulation of baseline heart rate in the fetal lamb. Am J Obstet Gynecol. 1992 Aug;167(2):519-23.
- Kleinhout J, Stolte LA, Janssens J, Knoop AA. The fetal autonomic nervous system, the fetal heart rate and the beat-to-beat irregularity. Eur J Obstet Gynecol Reprod Biol. 1977;7(6):373-6.
- Lear CA, Galinsky R, Wassink G, Mitchell CJ, Davidson JO, Westgate JA, Bennet L, Gunn AJ. Sympathetic neural activation does not mediate heart rate variability during repeated brief umbilical cord occlusions in near-term fetal sheep. J Physiol. 2016 Mar 1;594(5):1265-77.
- National Institute of Child Health and Human Development Research Planning Workshop. Electronic fetal heart rate monitoring: research guidelines for interpretation. Am J Obstet Gynecol. 1997 Dec; 177(6): 1385-90.
- Macones GA, Hankins GD, Spong CY, Hauth J, Moore T. The 2008 National Institute of Child Health and Human Development workshop report on electronic fetal monitoring: update on definitions, interpretation, and research guidelines. Obstet Gynecol. 2008 Sep; 112(3):661-6.
- ACOG Practice Bulletin No. 106: Intrapartum fetal heart rate monitoring: nomenclature, interpretation, and general management principles. American College of Obstetricians and Gynecologists. Obstet Gynecol. 2009 Jul; 114(1):192-202.
- American College of Obstetricians and Gynecologists. Practice bulletin no. 116: Management of intrapartum fetal heart rate tracings. Obstet Gynecol. 2010 Nov;116(5):1232-40.
- Liston R, Crane J, Hamilton E, Hughes O, Kuling S, MacKinnon C, McNamara H, Milne K, Richardson B, Trépanie MJ; Working Group on Fetal Health Surveillance in Labor, Executive and Council, Maternal-Fetal Medicine Committee, Clinical Practice Guideline Committee, and ALARM Committee, Society of Obstetricians and Gynaecologists Canada; Canadian Medical Protection Association. J Obstet Gynaecol Can. 2002 Mar;24(3):250-76.
- NICE Clinical Guideline 55. Intrapartum care http://www.nice.org.uk/nicemedia/live/11837/36273/36273.pdf accessed February 28, 2014.
- Parer JT, Ikeda T. A framework for standardized management of intrapartum fetal heart rate patterns.Am J Obstet Gynecol. 2007 Jul;197(1):26.e1-6.
- Clark SL, Nageotte MP, Garite TJ, Freeman RK, Miller DA, Simpson KR, Belfort MA, Dildy GA, Parer JT, Berkowitz RL, D’Alton M, Rouse DJ, Gilstrap LC, Vintzileos AM, van Dorsten JP, Boehm FH, Miller LA, Hankins GD. Intrapartum management of category II fetal heart rate tracings: towards standardization of care. Am J Obstet Gynecol. 2013 Aug; 209(2):89-97.
- Dalton KJ, Dawes GS, Patrick JE. Diurnal, respiratory, and other rhythms of fetal heart rate in lambs. Am J Obstet Gynecol. 1977 Feb 15; 127(4): 414-24.
- Ikenoue T, Martin CB Jr, Murata Y, Ettinger BB, Lu PS. Effect of acute hypoxemia and respiratory acidosis on the fetal heart rate in monkeys. Am J Obstet Gynecol. 1981 Dec 1; 141(7):797-806.
- Bocking AD. The relationship between heart rate and asphyxia in the animal fetus. Clin Invest Med. 1993 Apr; 16(2):166-75.
- Field DR, Parer JT, Auslender R, Baker BW, Ross BK, Leicht CH. Fetal heart rate variability and cerebral oxygen consumption in fetal sheep during asphyxia. Eur J Obstet Gynecol Reprod Biol. 1991 Nov 26;42(2):145-53.
- Low JA, Victory R, Derrick EJ. Predictive value of electronic fetal monitoring for intrapartum fetal asphyxia with metabolic acidosis. Obstet Gynecol. 1999 Feb; 93(2):285-91.
- Williams KP, Galerneau F. Intrapartum fetal heart rate patterns in the prediction of neonatal acidemia. Am J Obstet Gynecol. 2003 Mar; 188(3):820-3.
- Samueloff A, Langer O, Berkus M, Field N, Xenakis E, Ridgway L. Is fetal heart rate variability a good predictor of fetal outcome? Acta Obstet Gynecol Scand. 1994 Jan;73(1):39-44.
- Cahill AG, Roehl KA, Odibo AO, Macones GA. Association and prediction of neonatal acidemia. Am J Obstet Gynecol. 2012 Sep; 207(3):206. e1-8.
- Clark SL, Hamilton EF, Garite TJ, Timmins A, Warrick PA, Smith S. The limits of electronic fetal heart rate monitoring in the prevention of neonatal metabolic acidemia. Am J Obstet Gynecol. 2017 Feb;216(2):163.e1-163.e6
- Cahill AG, Tuuli MG, Stout MJ, López JD, Macones GA. A prospective cohort study of fetal heart rate monitoring: deceleration area is predictive of fetal acidemia. Am J Obstet Gynecol. 2018 Feb 1.
- Cyr RM, Usher RH, McLean FH. Changing patterns of birth asphyxia and trauma over 20 years. Am J Obstet Gynecol. 1984 Mar 1; 148(5):490-8.
- Hull J, Dodd KL. Falling incidence of hypoxic-ischaemic encephalopathy in term infants. Br J Obstet Gynaecol. 1992 May; 99(5):386-91.
- Smith J, Wells L, Dodd K. The continuing fall in incidence of hypoxic-ischaemic encephalopathy in term infants. BJOG. 2000 Apr; 107(4):461-6.
- Bell R, Glinianaia SV, Rankin J, Wright C, Pearce MS, Parker L. Changing patterns of perinatal death, 1982-2000: a retrospective cohort study. Arch Dis Child Fetal Neonatal Ed. 2004 Nov;89(6):F531-6.
- Becher JC, Stenson BJ, Lyon AJ. Is intrapartum asphyxia preventable? BJOG. 2007 Nov;114(11):1442-4. Epub 2007 Sep 17.
- Pasupathy D, Wood AM, Pell JP, Fleming M, Smith GC. Rates of and factors associated with delivery-related perinatal death among term infants in Scotland. JAMA. 2009 Aug 12;302(6):660-8.
- Lee ACC, Kozulki N, Blencowe H et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Ped Research 2013;74: 50-72.
- MacDonald D, Grant A, Sheridan-Pereira M, Boylan P, Chalmers I. The Dublin randomized controlled trial of intrapartum fetal heart rate monitoring. Am J Obstet Gynecol. 1985 Jul 1;152(5):524-39.




